
Research
Comparative Phylogeography and Population Genomics
A core theme of my research is to investigate how species have responded to environmental change in the past and whether they have the capacity to adapt to changing environments in the future.
I study genetic variation across geographic space (phylogeography) in a comparative framework to determine whether species in the same region are influenced by historical processes and contemporary landscape in similar ways, or whether species-level differences (e.g., dispersal ability) play an important role. My work in the Southeastern U.S. Coastal Plain sampled genome-scale data to quantify concordance across four widely-distributed frog species, and found that they exhibit largely discordant genetic patterns. Based on paleoclimate niche models, these four species are predicted to have responded differently to historical climate change, and these differences may be linked to species traits (e.g., the two more generalist species had higher niche stability). Currently, we are repurposing existing genetic data for >100 amphibian species in North America to ask whether species traits can predict within-species genetic diversity. This work is being conducted in collaboration with two Carstens-lab graduate students, Emanuel Fonseca and Cole Thompson.
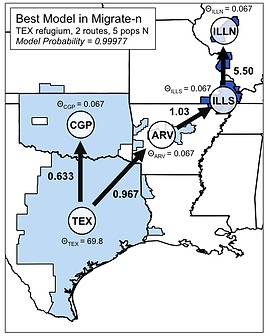
We are also actively collecting genome-wide SNP data (e.g., ddRAD) for several population-level studies. Undergraduate student Ivy Larkin is working with me to collect data from a dozen squamate species sampled in Florida to compare mitochondrial and nuclear genomic variation, and I am collecting a similar population genomic dataset for several frog species. These types of datasets enable comparison of complex demographic models, such as in this project with Alyssa Hassinger, one of my former undergraduates, in which we inferred post-glacial expansion routes and population connectivity in a chorus frog species complex.





Evolution and Ecology of Host-Parasite Interactions
Changes in species distributions lead to novel interactions between species. Such range shifts are expected to occur with increasing frequency because of climate change and the introduction of invasive species in our increasingly globalized world. Novel interactions with parasites can be particularly detrimental for hosts that do not have prior experience and lack defenses. One aspect of my research focuses on describing the abundance, diversity, and host associations of poorly-documented parasites to better understand the potential for these interactions. For example, this study led by Rosario Marroquin-Flores, a post-baccalaureate scholar I mentored, describes the results from our first year surveying for avian malaria and related haemosporidians across bird communities in New Mexico. A fantastic team of students, led my myself and Chris Witt (Museum of Southwestern Biology, University of New Mexico), nearly quadrupled our sampling to quantify parasite community turnover across three mountain ranges (manuscript in preparation).
Multi-host, multi-parasite assemblages such as avian haemosporidians also provide a fantastic system for asking a variety of questions in evolution and ecology. Why are some hosts more susceptible to parasites than others? How do some parasites infect multiple host while others are highly specialized on a single host? What is the geographic scale of turnover among parasite communities and how does this relate to host communities? I am collaborating with Staffan Bensch at Lund University, Sweden, to investigate phylogenomic relationships, species boundaries, and host range limits of avian haemosporidians using recently-developed genomic sequence capture methods. I have also recently initiated surveys of hemoparasites and potential pathogens of amphibian and reptile communities to begin uncovering the diversity and distributions of these little known, but potentially influential organisms (e.g., see Isidoro-Ayza et al. 2017; Karwacki et al. 2018).
Integrating Genomics and Natural History Collections
Museum collections are essential for understanding how species respond to environmental change through time. Perhaps the most captivating aspect of collections research is that ever-improving technologies unlock new information from specimens that the original collectors may never have anticipated. One example is the ability to compare the genetics of populations sampled at different timepoints to understand changes in diversity or how hybrid zone dynamics may have shifted, such as this project by Kristin Engebretsen, one of my former undergraduate mentees. In that case, D. Gartside had the foresight to store frozen tissues since 1976, which were then safely preserved by the Louisiana State University Museum of Natural Science. Genetic analysis of even older historical specimens can also be used to reconstruct phylogeographic histories of difficult-to-sample or even extirpated populations (e.g., Soto-Centeno et al. 2013, Barrow et al. 2016).
Another avenue of my research is to investigate host-pathogen interactions and parasite species limits by applying new genomic techniques to host tissues or specimens (e.g., Barrow et al. 2019). Parasite diversity is vastly under-described, and museum specimens preserve a wealth of information about when, where, and in what hosts disease-causing parasites occur.

Marker Development and Conservation Applications
Estimating demographic parameters such as population size or gene flow, inferring robust phylogenetic relationships, and testing complex historical models often benefit from genome-scale, multi-locus datasets. I have contributed to the development of new genetic tools that have not been readily available for the taxa I work on. Until recently, this work required huge numbers of PCR reactions, such as this project by myself and then-undergraduate Hannah Ralicki that generated a species tree hypothesis for North American chorus frogs using parallel tagged amplicon sequencing. Now I often use more broadly-applicable tools such as sequence capture or restriction-based methods for genome-scale projects.
Other genetic marker development projects have focused on species of conservation concern, such as the Illinois chorus frog, which occurs in three disjunct regions of the central U.S. where habitat loss and agricultural practices have led to population declines. We analyzed these genetic data in a Bayesian model comparison framework, which showed that disjunct clusters represent genetically distinct units and suggest they be managed as such. I also assisted with conservation-oriented marker development projects on species such as the endangered Pickersgill's Reed Frog and the blue and black wildebeest during a 6-month collaborative fellowship with researchers at the National Zoological Gardens of South Africa. I look forward to continued local and international collaborations with conservation professionals seeking to expand their genetic resources.

